
Proximal muscle weakness (BMJ Practical neurology)
Pract Neurol 2019;19:321–325.
DOI:10.1136/practneurol-2019-002204
近位筋筋力低下
タイトルは普通だな,と思って読みましたが,内容は非常に興味深い内容でした.
希少疾患ではありますが,治療の可能性がある疾患として重要であると感じました.国内ではどのように検査すればいいか……また調べたいと思います.
※一部,本邦で保険適用外の治療法が記載されています.ご留意ください.
症例
38歳 女性
【現病歴】
1年前から階段を上ることや長距離歩行が難しくなり,車椅子が必要になった.
最近になって,上肢の筋力低下も出現し,髪を結ぶことが困難になり,軽い物も持てなくなった.軽労作で息切れするようになり,筋萎縮で体重が12ポンド(5.4kg)減った.
筋痛や褐色尿,筋痙攣,視覚障害,聴力障害,構音障害,嚥下障害,感覚低下,異常感覚,膀胱直腸障害,発熱,皮疹,関節痛などはなかった.成長発達は正常であった.
【既往歴】
特記すべき既往はない.
脂質降下薬は使用しておらず,違法薬物使用や毒物への曝露はない.
【家族歴】
家族歴はなく,血族婚もない.
【診察所見】
両側性で左右対称性の翼状肩甲を認める.
徒手筋力試験では,頸部屈曲4-,頸部進展5,肩関節外転4/4,肘外旋4/4,肘屈曲4/4,肘伸展4+/4+,手首屈曲5/5,手首伸展5/5,手指屈曲5/5,手指伸展5/5,股関節屈曲4/4,股関節伸展3/3,股関節外転3/3,膝伸展4+/4+,膝屈曲4/4,足背屈5/5,足関節底屈5/5.
腱反射は,三角筋,腕撓骨筋,アキレス腱で1+,上腕二頭筋,膝蓋腱で2+.
歩行は動揺性.足底反射は屈曲.感覚や協調運動は正常.
把握ミオトニアやパラミオトニア,線維束攣縮は認めない.
筋肉量やトーヌスは正常.脳神経は正常.
Question 1:鑑別疾患は?
対称性な上下肢の近位筋筋力低下はミオパチーを考える.
鑑別疾患として,後天性ミオパチー(例:炎症性,中毒性ミオパチー),遺伝性ミオパチー(例:筋ジストロフィー,遅発性先天性ミオパチー/代謝性ミオパチー)など.
眼神経筋接合部疾患も考慮する必要がある(筋無力症の90%で眼筋症状がある).筋症状や症状の変動はない点は重症筋無力症としては非典型的で,自律神経症状が無い点と,膝蓋腱反射が保たれていることはLambert-Eaton症候群として非典型的である.
SMA typeⅢなどの運動ニューロン疾患は本例よりも緩徐に進行する.
Question 2: 検査は何を行うか?
血清CK 682~1464 U/Lと上昇.
血算,電解質,抗核抗体,ANCA,TSH,赤沈,CRP,血清蛋白免疫固定法,B型肝炎検査,C型肝炎検査,筋炎特異的抗体パネルは,すべて正常あるいは陰性.
筋電図は右胸部脊柱起立筋と右腓腹筋で自発放電(Positive sharp waveとFibrillation)を認めるが,MUP形態異常や動員に異常は認めない.
右腓腹筋の筋生検は正常.
経胸壁心エコーは正常.
Question 3:検査結果をどのように解釈する?
Positive sharp waveとFibrillationは神経原性でも筋原性でも生じうる非特異的な所見である.MUP形態異常と動員所見は神経原性と筋原性の鑑別に役立つが(神経原性でhigh amplitudeとlong durationで動員低下.筋原性でlow amplitudeとshort durationで早期動員),本症例では見られなかった.
生検に適した筋は,臨床的に筋力低下しているが廃絶しきっていない筋である.本例では右腓腹筋は筋力が正常であり,筋生検が診断につながらなかったと考えた.
本症例では,病歴や診察所見,高CK血症などから,筋疾患を考える.
考えられる筋疾患として,成人発症の腰肢帯型筋ジストロフィーを最も考える(翼状肩甲は後天性ミオパミーでは稀).
それよりも頻度は低くなるが、幾つかの先天性ミオパミー(例:セントラルコア)は,本例のように成人発症する.
代謝性ミオパミーも考慮すべきである.本例はMcArdle病のような運動不耐症やミオグロビン尿症はない.しかし,成人発症Pompe病は進行性近位筋筋力低下や,軽度~中等度のCK上昇,筋電図での筋原性変化を呈する.Pompe病歴は現在,酵素補充療法があるため,考慮する事が重要である.
稀だが,脂肪蓄積性ミオパミーも同様の臨床像を呈する.しかし,急速に進行する脂肪蓄積性ミオパチー(ミトコンドリア三頭酵素欠損症,極長鎖アシル CoA 脱水素酵素欠損症,カルニチンパルミトイルトランスフェラーゼ-Ⅱ欠損など)らしくはない.横ばい~進行性で経過する脂肪蓄積性ミオパチーは考慮すべきである.
Question 4: どのように検査を続けるべきか?
Pompe病のスクリーニングとして,血清アルファグルコシダーゼ活性を行ったが異常なし.
腰肢帯型筋ジストロフィーで多い遺伝子変異(ANO5, CAPN3, CAV3, DMD, DES, DNAJB6, DYSF, FKRP, FKTN, GAA, GMPPB, LMNA, MYOT, POMGNT1, POMT1, POMT2, SGCA, SGCB, SGCD, SGCG, TCAP, TNPO3, TRIM32 , TTN)も検査したが,異常はなかった.
左上腕二頭筋と左三角筋で筋電図を施行し,刺入時活動の増加と,自発放電(Positive sharp waveとFibrillation),早期動員,short-duration, low-amplitude,多相性のMUPを認めた.
同日,右三角筋の筋生検を施行.

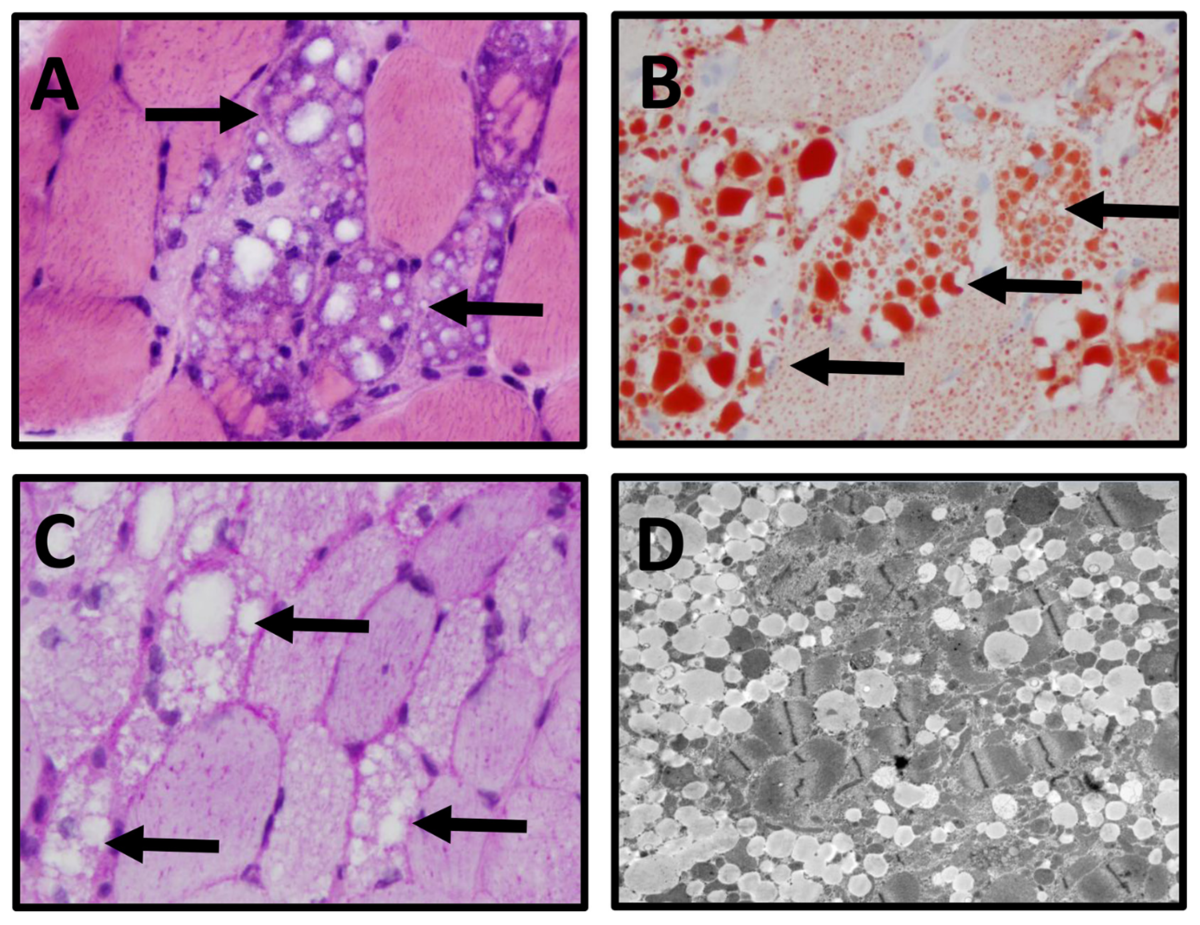
A:筋繊維の空胞化(HE染色)
B:細胞質内に脂肪を含んだ小胞(oil red O染色)
C:グリコーゲンは認めない(PAS染色)
D:筋繊維内に多数の脂肪小胞(電子顕微鏡)
Question 5: 確定診断に最も有用な検査は?
本症例は,広範に筋繊維内の脂肪蓄積を認め,脂肪蓄積性ミオパチーと考えられた.
脂肪蓄積性ミオパチーは,原発性カルニチン欠損症や,複合アシルCoA脱水素酵素欠損症(MADD),中性脂質蓄積症)などで生じる.これらの疾患は,運動不耐症より,むしろ進行性の近位筋筋力低下をきたす.
血清遊離カルニチン,血清総カルニチン,血清アシルカルニチン(絶食12時間の検査が診断に有用),尿中有機酸がこれら3つの鑑別に有用である.
原発性カルニチン欠乏症では,血清遊離&総カルニチン,血清アシルカルニチンが著明に低下するが,尿中有機酸は正常である.
複合アシルCoA脱水素酵素欠損症(MADD)では,血清の短鎖(C4-8),中鎖(C8-12),長鎖(>C12)アシルカルニチンは上昇し,血清遊離&総カルニチンは正常から二次的に低下する,尿中有機酸は上昇する.
中性脂肪蓄積病では,蓄積するのは脂肪酸ではなく,トリグリセライドであるため,血清血清遊離&総カルニチンと血清アシルカルニチン,尿中有機酸はすべて正常である.

本例の血清遊離カルニチン濃度は 19 μmol/L(正常:25~60) と軽度低下し,総カルニチンは 23 μmol/L(正常:5~29)と正常範囲であった(原発性カルニチン欠乏症は除外).アシルカルニチン分画では,短・中・長鎖アシルカルニチン濃度は上昇し,尿中有機酸では2-hydroxyglutaric acidが上昇した.
これらの所見からMADDが示唆された.
Question 6: どのように確定診断し治療するか?
MADDは常染色体劣性遺伝の疾患で,93%がETFDHの変異で生じる(ETFDHはelectron transfer flavoprotein–ubiquinone oxidoreductase (ETF-QO)をコードする).多くは,リボフラビン投与に反応する.
本例のETFDH遺伝子検査で,c.814G>A, p.Gly272Arg と c.1204A>G, p.Thr402Alaの2つの変異を認めた.
3週間のリボフラビン内服(400mg/日)で筋力と身体機能は著明に改善した.車椅子なしであるけるようになり,歩行時の息切れが無くなり,手の力を使わず椅子から立ち上がることができるようになった.3ヶ月後,筋力は改善した:頸部屈曲4+,肩外転5/5,肘外旋5/5,肘屈曲5/5,肘伸展5/5,股関節伸展4/4,股関節外転4/4,膝伸展5/5.膝屈曲5/5.治療3ヶ月時点でCKは89 U/Lで,血清アシルカルニチンは正常化~低下した.
リボフラビン400mg/日を継続することとした.
コメント
MADD(グルタル酸尿症2型とも呼ばれる)は常染色体劣性遺伝形式の疾患で,STFDH変異が93%,ETFA/B変異が(~7%)が原因である.ミトコンドリア内での短鎖,中鎖,長鎖脂肪酸から呼吸鎖へのβ酸化での電子の受け渡しが阻害される.結果的に,すべての長さの脂肪酸が増える.血清ではアシルカルニチンの形で,尿では有機酸の形で増え,それらは脂肪酸酸化の障害を示唆する.
リボフラビン(ビタミンB2)はフラビンアデノシンジヌクレオチドの前駆体で,EETFDH(ETF-QO)とETFA/B(ETF)でコードされるフラボプロテインの補因子として必要である.リボフラビン100~400mg/日投与が効果があったとするMADDが少数報告されている.
MADDの表現形は年齢により大きく異なる.成人期発症例は,近位筋ミオパチーを呈する.新生児発症例では,しばしば多臓器不全で致死的な経過をたどる.
CKや筋電図,血清アシルカルニチン分画,尿有機酸が正常であってもMADDは除外できない.非特異的だが,MRIで大腿部の背側や臀部の筋での脂肪置換や筋萎縮がみられる.MADDの診断は筋生検と遺伝子検査が主である.筋生検では,広範な筋肉内脂肪滴蓄積を認める.
本症例のミオパチーは,多くの場合リボフラビン投与が著効する.〈本邦では保険適応外〉
素敵なブログですね!読者登録させて頂きました✨
ありがとうございます!😊